Over the past twenty years there has been a dramatic change in approach to the treatment of heart failure. We have progressed from an era where the only drugs available were digoxin and diuretics, to an era where multiple new drugs with different strategies are becoming available. The development of new drugs is partly related to the explosion in our knowledge of the processes involved in heart failure. It is essential that the clinician understand the principal mechanisms underlying congestive heart failure for logical selection of treatment.
Pathophysiology of heart failure
The traditional theory of heart failure basically viewed the dysfunction as a 'plumbing' problem. When the heart failed as a pump, the tissues (including the kidneys) were inadequately supplied with blood, leading to reduced renal function and sodium and water retention. The combination of sodium and water retention, together with a backwards "damning back" of pressure resulted in the signs of congestive heart failure. The traditional approach was to treat congestive heart failure by improve pump function with digoxin, and removing excess sodium & water with diuretics.
The contribution of autonomic reflexes and production of neurohormones in the development of heart failure is now widely recognized. The cardiovascular system is regulated by a series of inter-related neurohormonal responses. The major goal of homeostasis for the cardiovascular system is maintenance of arterial blood pressure, which must be tightly controlled to ensure adequate cerebral and coronary perfusion. 'Compensatory mechanisms' are brought into play in an effort to maintain arterial blood pressure at an adequate level, irrespective of the underlying cause of fall in blood pressure. These compensatory mechanisms are very successful in maintaining blood pressure, but can have adverse hemodynamic effects on the heart by increasing vascular resistance and therefore afterload, and by increasing myocardial oxygen consumption.
Arterial "under-filling" triggers a change in baroreceptor activity which increases sympathetic outflow. The increase in cardiac output produced by increased contractility and heart rate helps to restore normal arterial blood pressure. At the same time, the increased systemic vascular resistance also raises arterial pressures. However, both the increased cardiac output and the vasoconstriction result in increased myocardial oxygen consumption, i.e., the heart has to work harder, at higher energy cost. Additional problems associated with high circulating catecholamine levels include an increased risk of ventricular arrhythmias as a result of increased cytosolic calcium levels.
Neurohormones in Heart Failure
"Harmful"
 Noradrenaline
Noradrenaline
Adrenaline
Angiotensin II
Aldosterone
Arginine vasopressin
Endothelin
TNFalpha
"Beneficial"
Acetylcholine
ANP
BNP
NO
Bradykinin
Cardiovascular adrenergic effects
ß cardiac effects

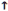 contractility
contractility
heart rate
relaxation
alpha 1 vascular effects
Vasoconstriction
ß vascular effects
Vasodilation
Renin-Angiotensin-Aldosterone system
The combination of increased sympathetic drive with reduced renal perfusion pressures leads to release of renin from the juxtaglomerular apparatus, which converts angiotensinogen to angiotensin I. Angiotensin converting enzyme (ACE) converts angiotensin I to angiotensin II. Angiotensin II has many effects, most of which contribute to vasoconstriction or sodium and water retention. Arterial vasoconstriction increases arterial pressures but increases afterload, and sodium and water retention increases preload and augments cardiac output but increases filling pressures.
Other neurohormones
Arginine vasopressin is a potent vasoconstrictor and also induces free water retention. Release of vasopressin occurs in response to arterial under-filling or increased sympathetic drive. Endothelin causes vasoconstriction, and levels are also increased in heart failure.
Overview of neurohormonal activation
Arterial under-filling leads to production of neurohormones that cause vasoconstriction and/or sodium and water retention, both of which restore normal arterial pressures but at increased energy cost to the myocardium. Baroreceptor sensitivity is decreased in heart failure, so that although normal arterial pressures are restored, increased sympathetic drive continues. Although heart failure also increases concentrations of some 'beneficial' neurohormones (atrial natriuretic peptide, brain natriuretic peptide), the vasodilatory and natriuretic properties of these hormones tends to be overwhelmed by the effects of the neurohormones mentioned above. An understanding of the importance of neurohormonal mechanisms in heart failure has resulted in some changes to our approach in treatment. Diuretics are still employed for elimination of sodium and water, but ACE inhibitors have assumed increasing importance. Digoxin is now used as much for its effects in restoring baroreceptor sensitivity as for its positive inotropic effects.
Ventricular remodeling
Further refinements to the neurohormonal theory of heart failure have shown that there are adverse effects on the myocardium in addition to the increased workload imposed by neurohormonal activation. These are local tissue effects, whereby the structure and function of the myocardium itself is altered by neurohormones. In vivo, catecholamines contribute to myocyte loss. There are a number of mechanisms that attempt to limit the effect of catecholamines on the myocardium, such as beta-receptor down-regulation. Other neurohormones also have direct tissue effects: angiotensin II and aldosterone promote interstitial fibrosis. The net result is a decrease in diastolic and systolic function attributable to the effects of neurohormones, independent of the initial cardiac lesion. The heart dilates, and becomes less efficient mechanically, leading to further worsening of cardiac function and further neurohormonal activation. However, therapy directed at reducing remodeling (e.g., beta-adrenergic antagonists) sometimes has adverse short-term hemodynamic effects. Therapy aimed at reducing neurohormonal activation in general will be expected to help reduce progression of ventricular remodeling.
Implications for treatment of heart failure
The aims of acute therapy of heart failure are clearly different from the therapy of chronic heart failure. Acute therapy must concentrate on preventing death from hypoxia, and improving hemodynamics. The effects on neurohormones are not so important in the short-term.
The goal of chronic therapy is to remove excess sodium and water retention, and modulating harmful neurohormones so that congestive signs are less likely. Ideally, therapy should help to limit the effects of ventricular remodeling. Drugs that result in neurohormonal activation (dobutamine, hydralazine) are best avoided. However, drugs that directly limit sympathetic drive (i.e., beta-adrenergic antagonists) are difficult to use, because they cause a direct fall in cardiac output in the short-term and are poorly tolerated. Their use has not been studied in naturally occurring heart disease in dogs. An alternative approach is to use pimobendan to increase cardiac output, which rather than causing neurohormonal activation, may actually decrease neurohormones indirectly.
Acute Therapy
Treat hypoxia
Oxygen
Drain pleural effusions
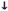 Atrial pressures
Atrial pressures
Furosemide
Venodilators (nitroglycerine)
Cardiac output
(Dobutamine)
Pimobendan
Chronic Therapy
Na+ and H2O retention
Furosemide
Neurohormones
ACE inhibitors
Spironolactone
? Digoxin
Cardiac output
Digoxin
Pimobendan
Newer Heart Failure Therapy
Pimobendan
Most positive inotropes increase the cytosolic concentration of calcium to increase the availability of calcium, but pimobendan has the novel action of increasing the sensitivity of the contractile proteins to calcium, an effect thought to be mediated by altering the binding of calcium to the troponin complex, and the increase in the extent of sarcomere shortening is achieved without the same energy consumption associated with sympathomimetic drugs. Pimobendan has both calcium sensitizing effects and some phosphodiesterase inhibitory effects. The calcium sensitizing effects appear to predominate in failing myocardium, because of down-regulation of the adrenergic signaling pathway. The phosphodiesterase inhibition also results in vasodilation. Phosphodiesterases are involved in the breakdown of cAMP in vascular smooth muscle, and inhibitors of these phosphodiesterases will cause venodilation and arteriodilation. There has been no significant increase in mortality in clinical human studies, and neurohormonal activation appears to be reduced with its use.
Pimobendan is available as an oral preparation, and can be used in both acute and chronic heart failure. In acute heart failure it can be used in place of positive inotrope infusions, and will increase cardiac output and decrease filling pressures. Normally hypotension is not a problem because of concurrent augmentation of contractility, although blood pressure should be monitored. At normal dose rates of 0.1-0.3 mg/kg q12 hours it should not affect heart rate and is not arrhythmogenic. In chronic heart failure, it can be used long-term to increase contractility without causing neurohormonal activation.
Spironolactone
Spironolactone is not a new treatment, but its use has recently been reevaluated in human heart failure patients. Once thought of only as a potassium-sparing diuretic, it is now viewed as a useful adjunct to countering the renin-angiotensin-aldosterone system. Aldosterone not only has adverse cardiovascular effects by leading to sodium and water retention, but also has directly harmful myocardial tissue effects.
Beta-adrenergic antagonists
Attitudes towards beta-blockade in the field of human dilated cardiomyopathy have undergone dramatic changes in the past 10 years. Beta-adrenergic blockers have traditionally been considered to be contraindicated in myocardial failure, because of their adverse acute hemodynamic effects. It now appears that long-term use of beta-adrenergic antagonists (>3 months) is associated with improvement in systolic function in human patients, an effect which seems paradoxical, as the short-term hemodynamic effects are negatively inotropic. It was initially believed that the mechanism was related to reversal of the down-regulation of ß-receptors that occurs with chronically elevated catecholamine levels; however, some beta-adrenergic blockers produce an improvement in systolic function without causing any ß-receptor up-regulation. It now appears that the improvement is related to an increase in contractile function in the cardiac myocytes themselves, which may be a result of an increase in contractile elements. Certain strict guidelines are recommended for commencing therapy with beta-adrenergic antagonists in human patients with myocardial failure: patients must be stable and compensated (i.e., no congestive signs); doses must be extremely low initially (often < 0.1 of target dose); and doses must be titrated upwards slowly (at 1-2 week intervals). The introduction of beta-blockers with concurrent alpha-1 adrenergic blocking effects (such as carvedilol) has led to improved tolerance in heart failure patients, as the vasodilatory effects offset the decrease in stroke volume associated with the negative inotropic effects.
To date, there have been no reported studies of the effects of beta-blockade on systolic function in dogs with naturally occurring heart disease, although one study in dogs with experimental mitral regurgitation did show an improvement in contractile function of isolated cardiomyocytes following atenolol therapy. In the absence of further data, it is speculative whether beta-blockers will eventually have a place in treatment of canine myocardial failure. Problems with canine patients include the long titration period required before beneficial effects might be seen: unless the projected survival time exceeds 3 months, there is little point in starting a beta-blocker for its long term effects. Furthermore, human patients often report feeling worse before they feel better with beta-blockers, and this short-term effect may be difficult to justify in dogs with DCM. At present, the use of beta-blockers in canine DCM is certainly indicated for heart rate control in atrial fibrillation (in conjunction with digoxin) or antiarrhythmic therapy for ventricular arrhythmias, but there are no recommended guidelines for use in dogs with DCM and controlled heart failure. Clinical studies are currently underway to investigate use of carvedilol in canine heart failure.