Abstract
Selecting the correct antibacterial drug is one of the most challenging therapies encountered in veterinary medicine. Optimum selection should be based on a logical, systematic approach. This paper will focus on some of the important strategies that can guide antibacterial drug selection in small animal patients.
Penetration to the Site of Infection
For most tissues, antibiotic drug concentrations in the serum or plasma approximate the drug concentration in the extracellular space (interstitial fluid). This is because there is no barrier that impedes drug diffusion from the vascular compartment to extracellular tissue fluid.1 Pores in the endothelium of capillaries are large enough to allow drug molecules to pass through unless the drug is highly protein bound in the blood. Fortunately, there are no antibiotics that have been shown to have high drug protein binding in the plasma to the extent that it is clinically relevant.
Diffusion of most antibiotics in tissues is limited by tissue blood flow, rather than drug lipid solubility. This has been called perfusion-rate limited drug diffusion. If adequate drug concentrations can be achieved in plasma, it is unlikely that a barrier in the tissue will prevent drug diffusion to the site of infection, as long as the tissue has an adequate blood supply. Drug diffusion into an abscess or granulation is sometimes a problem because these conditions lacks adequate blood supply and drug penetration relies on simple diffusion. Low drug concentrations in an abscess also is possible because in a cavitated lesion there is low surface area to volume ratio (low S:V ratio) rather than a physical barrier to diffusion.
In some tissues a lipid membrane (such as tight junctions on capillaries) presents a barrier to drug diffusion. This has been called permeability-rate limited drug diffusion. In these instances, a drug must be sufficiently lipid-soluble, or be actively carried across the membrane in order to reach effective concentrations in tissues. These tissues include: the central nervous system, eye, and prostate. There also is a barrier between plasma and bronchial epithelium. This limits drug concentrations of some drugs in the bronchial secretions and epithelial fluid of the airways, but not to the interstitial fluid of the lung. To achieve effective drug concentrations in bronchial secretions, drugs must be sufficiently lipid soluble to diffuse through the lipid barrier.
Intracellular Infections
Most bacterial infections are located extracellularly, and a cure can be achieved with adequate drug concentrations in the extracellular (interstitial) space rather than intracellular space. Intracellular infections present another problem. For drugs to reach intracellular sites, they must be carried into the cell or diffuse passively. For passive diffusion to occur, only lipid-soluble drugs will be able to diffuse through the cell membrane. Intracellular organisms such as Brucella, Chlamydia, Rickettsia, Bartonella, and Mycobacteria are examples of obligate intracellular pathogens. Staphylococci may in some cases become resistant to treatment because of intracellular survival. Fluoroquinolones and tetracyclines such as doxycycline are frequently administered to treat Rickettsia and Ehrlichia infections. Examples of drugs that accumulate in leukocytes and other cells are fluoroquinolones, lincosamides (clindamycin, lincomycin), macrolides (erythromycin, clarithromycin), and the azalides (azithromycin).2 Beta-lactam antibiotics and aminoglycosides do not reach effective concentrations within cells.
Local Factors that Affect Antibiotic Effectiveness
Local tissue factors may decrease antimicrobial effectiveness. For example, pus and necrotic debris may bind and inactivate vancomycin or aminoglycoside antibiotics (for example, gentamicin or amikacin), causing them to be ineffective. Foreign material in a wound (such as material surgically implanted) can protect bacteria from antibiotics and phagocytosis by forming a biofilm (glycocalyx) at the site of infection. Cations such as calcium and magnesium can adversely affect the activity of antimicrobials at the site of infection. Two important groups diminished in activity by cations are fluoroquinolones and aminoglycosides.
An acidic environment of infected tissue may decrease the effectiveness of clindamycin, erythromycin, fluoroquinolones, and aminoglycosides. Penicillins and tetracycline activity is not affected as much by tissue pH, but hemoglobin at the site of infection will decrease the effectiveness of these drugs. An anaerobic environment decreases the effectiveness of aminoglycosides, whereas metronidazole has no activity against aerobic bacteria.
As mentioned previously, an adequate blood flow is necessary to deliver an antibiotic to the site of infection. Effective antibacterial drug concentrations may not be attained in tissues that are poorly vascularized (e.g., extremities during shock, sequestered bone fragments, and endocardial valves).
Bacterial Susceptibility
If the bacteria is accurately identified, antibiotic selection is simplified because the susceptibility pattern of many organisms is predictable. For example, if the bacteria is likely to be Pasteurella, or Streptococcus, or Actinomyces, susceptibility is expected to penicillin or an aminopenicillin such as ampicillin, amoxicillin, or amoxicillin-clavulanic acid (Clavamox). Other susceptibility patterns are discussed below:
Staphylococcus intermedius
Staphylococcus isolated from small animals is most likely to be S. intermedius rather than S. aureus. S. intermedius will usually have a predictable susceptibility to β-lactamase resistant β-lactam antibiotics such as oxacillin or dicloxacillin, amoxicillin combined with a β-lactamase inhibitor, or a first-generation cephalosporin such as cephalexin or cefadroxil. Reports of studies on S. intermedius have shown that, despite frequent use of the above-mentioned drugs in small animals, the incidence of resistance has not increased.3
Anaerobes
If the bacteria is an anaerobe (for example, Clostridium, Fusobacterium, Prevotella, Actinomyces, or Porphyromonas), predictable results can be attained by administering a penicillin, chloramphenicol, metronidazole, clindamycin, amoxicillin-clavulanic acid, or one of the second-generation cephalosporins, such as cefoxitin. The activity of first-generation cephalosporins, trimethoprim-sulfonamides/ormetoprim-sulfonamides, or fluoroquinolones for an anaerobic infection is unpredictable.
Gram-Negative Bacilli
If the organism is Pseudomonas aeruginosa, Enterobacter, Klebsiella, E. coli, or Proteus, resistance to many common antibiotics is possible and a susceptibility test is advised. However, for initial therapy, we usually expect the gram-negative enteric bacteria to be susceptible to fluoroquinolones and aminoglycosides. An extended-spectrum cephalosporin (second- or third-generation cephalosporin) usually is active against enteric-gram-negative bacteria, but not Pseudomonas aeruginosa. If the organism is a Pseudomonas aeruginosa, inherent resistance to many drugs is common, but it may be susceptible to fluoroquinolones, aminoglycosides, or an extended-spectrum penicillin such as ticarcillin or piperacillin. Among the fluoroquinolones, ciprofloxacin is the most active against P. aeruginosa. In rare cases, one of the carbapenems such as imipenem is used to treat infections caused by resistant gram-negative bacteria.
Bacterial Susceptibility Testing
The minimum inhibitory concentration (MIC) of an antibiotic is the lowest concentration of the drug that inhibits visible bacterial growth. This is determined directly, by performing serial dilutions of the antibiotic and determining growth against each dilution. An indirect measurement of the MIC is the agar-disk-diffusion test (ADD). The ADD test results are only qualitative (that is, it determines only resistant vs. sensitive), but if performed using standardized procedures, this test is valuable.4
It is becoming more common for laboratories to directly measure the MIC for an organism.4 In this test, resistance and susceptibility are determined by comparing the organism's MIC to the drug's breakpoint. If bacteria have a MIC above the "resistant" breakpoint, the organism is resistant regardless of the dose administered or location of the infection. The MIC can be determined for each bacteria isolated. MIC tests are more quantitative than an ADD test, but must be performed according to strict guidelines.4
Pharmacokinetic–Pharmacodynamic Optimization of Doses
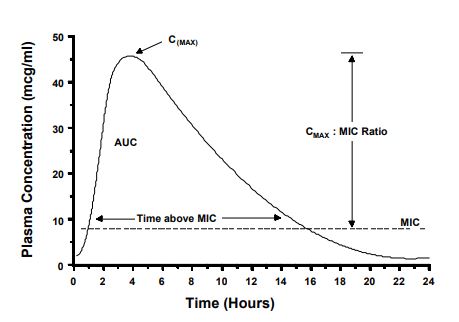
To achieve a cure, the drug concentration in plasma, serum, or tissue fluid should be maintained above the MIC, or some multiple of the MIC, for at least a portion of the dose interval. Antibacterial dosage regimens are based on this assumption. However, antibacterial drugs vary with respect to the peak concentration and the time above the MIC that is needed for a clinical cure. Pharmacokinetic/pharmacodynamic (PK-PD) relationships of antibiotics attempt to explain how these factors correlate with clinical outcome.5,6 Shown on Fig. 1 are some terms used to describe the shape of the plasma concentration vs. time profile. The CMAX is simply the maximum plasma concentration attained during a dosing interval. The CMAX is related to the MIC by the CMAX:MIC ratio. The AUC is the total area-under-the-curve. This measurement is taken from time zero to infinity after a single dose. The AUC is related to the MIC value by calculating the AUC:MIC ratio (also called the area-under-the-inhibitory curve [AUIC]). Also shown in Fig. 1, is the relationship of time to MIC measured in hours. Examples of how these relationships affect drug regimens are described below.
Figure 1. Terms used to describe the shape of the plasma concentration vs. time profile
Aminoglycosides
Aminoglycoside (e.g., gentamicin, or amikacin) are bactericidal when the peak plasma concentration are several times above the MIC. If a high enough dose is administered once daily it will produce a peak of 8–10× the MIC. This regimen is at least as effective, and perhaps less nephrotoxic than lower doses administered more frequently.7 Our regimens in small animals employ this strategy. Gentamicin has been administered safely and effectively at a dose of 7–10 mg/kg once daily, i.v. The corresponding dose for amikacin is 10–15 mg/kg, i.v.
Fluoroquinolones
For the fluoroquinolone antimicrobials, either the CMAX:MIC ratio, or the AUIC may predict antibacterial success. A peak concentration that is 8–10× the MIC, or a AUC:MIC ratio of 125 to 250 has been associated with the optimum antibacterial effect.8,9 In some immunocompetent animals, lower AUIC ratios that are 50–60 also may be effective. We administer doses to achieve high CMAX:MIC ratios whenever possible because high ratios have been associated with a lower incidence of development of resistance.6,9 To achieve this goal, low doses of fluoroquinolones (for example enrofloxacin 5 mg/kg/day) have been administered to treat susceptible organisms with low MIC, such as E. coli or Pasteurella, but for bacteria with a higher MIC (for example, gram-positive cocci), a larger dose will be needed. To achieve the necessary peak concentration for a bacteria such as Pseudomonas aeruginosa, that usually has the highest MIC among susceptible bacteria, the highest dose in a range is administered (for example, 15–20 mg/kg/day for enrofloxacin). When the MIC values are above the breakpoint, the effectiveness of fluoroquinolones is doubtful because even at high doses, a sufficient CMAX:MIC ratio or AUC:MIC ratio will be difficult to achieve.
Beta-lactam Antibiotics
β-lactam antibiotics such as penicillins, potentiated-aminopenicillins, and cephalosporins are slowly bactericidal. Their concentration should be kept above the MIC throughout most of the dosing interval.10 Dosage regimens for the β-lactam antibiotics should consider these pharmacodynamic relationships. Therefore, for treating a gram-negative infection, especially a serious one, administer penicillin derivatives and cephalosporins 3–6 times per day. Some of the third-generation cephalosporins have long half-lives and less frequent regimens have been used for some of these drugs (for example cefotaxime and ceftiofur). Gram-positive organisms are more susceptible to the β-lactams than are gram-negative bacteria. Additionally, since the MICs are lower for gram-positive bacteria, and antibacterial effects occur at concentrations below the MIC, longer dose intervals may be possible for infections caused by gram-positive as compared to gram-negative bacteria. For example, cephalexin or amoxicillin-clavulanate have been used successfully to treat staphylococcal infections when administered twice daily.
Bacteriostatic Drugs
The drugs such as tetracyclines, macrolides (erythromycin and derivatives), sulfonamides, lincosamides (lincomycin and clindamycin), and chloramphenicol derivatives act in a bacteriostatic manner. These drugs inhibit, but may not always kill the bacteria. Subsequently, the drug concentrations should be maintained above the MIC throughout the dosing interval. In this way, they act in a time-dependent manner. Most of the bacteriostatic drugs must be administered frequently to achieve this goal, but some have long half-lives or concentrations persist in tissue that maintain inhibitory drug concentrations throughout the dosing interval with infrequent administration (for example macrolides or trimethoprim-sulfonamides). Most dosage regimens are designed to take these drug’s pharmacodynamic action into account.
Update on Recent Developments in Drug Therapy
There have been several new developments in antibacterial therapy in the last ten years.11 Many of these changes affect drug therapy in zoo and exotic animals because some of these drugs have important uses in these species. Most of these developments have come from a need to treat resistant bacteria with safe drugs. There also is a need to find effective drugs that can be administered more conveniently. In this section is a brief summary of a few of the recent developments for drugs in these animals.
Cephalosporins
The 1st generation cephalosporins include common drugs such as cephalexin (Keflex), cefadroxil (Cefa-Tabs), and cefazolin. Cephalexin and cefadroxil are popular oral drugs; cefazolin is a common injectable antibiotic used for acute treatment and surgical prophylaxis. The spectrum of 1st generation cephalosporins includes most gram-positive cocci, including staphylococcus and some gram-negative bacilli. However, resistance among gram-negative Enterobacteriaceae is common. Extended-spectrum cephalosporins have been classified into the 2nd, 3rd, and 4th generation. The situations in veterinary medicine for which extended-spectrum cephalosporins are most often used are treatment of bacterial infections resistant to other drugs. The bacteria often implicated in resistance problems have been E. coli, Klebsiella pneumoniae, Enterobacter species, and Proteus species. Of the 2nd generation cephalosporins, cefoxitin and cefotetan are the most often administered in veterinary medicine. Their use has been valuable for treating organisms resistant to the 1st generation cephalosporins or in cases in which there are anaerobic bacteria. When anaerobic bacteria of the Bacteroides fragilis group become resistant, they produce a cephalosporinase. Cefoxitin and cefotetan are resistant to this enzyme and may be active against these bacteria. Therefore, these drugs may be valuable for some cases such as septic peritonitis that may have a mixed population of anaerobic bacteria and gram-negative bacilli.
The 3rd generation cephalosporins are the most active of the cephalosporins against gram negative bacteria, especially enteric bacteria resistant to other cephalosporins. Compared to other drugs in this group, cefotaxime and ceftizoxime have the best activity against staphylococci. Most of the drugs in this group, must be administered i.v. or i.m. (for convenience, some have been administered to animals s.c.). One should be aware that i.m. or s.c. administration of these drugs can be painful. Cefotaxime, ceftriaxone and ceftazidime are the only cephalosporins that can achieve adequate concentrations in the CSF and for this reason are a rational choice for treating septic meningitis. Compared to other cephalosporins, ceftazidime is the most active against Pseudomonas.
Cefixime has been examined in dogs because it is one of the only 3rd generation cephalosporins that can be administered orally. Ceftiofur (Naxcel®) fits most criteria for a 3rd generation cephalosporin, but also has activity against gram-positive bacteria and some have suggested that it should be in a separate class. Ceftiofur is a popular cephalosporin for use in cattle, but also has been used in pigs, sheep, horses, and dogs. In small animals it has been used for urinary tract infections caused by E. coli. In large animals it has been used primarily for respiratory infections. When comparing the 3rd generation cephalosporins, one should recall that a susceptibility test that indicates an organism is sensitive to one of the 3rd generation cephalosporins (for example cefotaxime), does not necessarily imply that the organism is sensitive to other 3rd generation cephalosporins.
There is little pharmacokinetic information for cephalosporins in animals other than domestic species. Fortunately, for most mammals, distribution and elimination are similar and dosage regimens do not vary much among species. Absorption differences occur and oral absorption is minimal in ruminants and horses. In non-mammals, some important differences have been identified that are most likely related to prolonged excretion from the kidneys. Elimination in reptiles is slower. For example, in a study of ceftazidime in sea turtles, we found that excretion is much longer and half-lives of 20 hr allow for dosing as infrequently as every 72 hr.12 Regimens for cephalosporins in other reptiles have been published.13 In birds, poor oral absorption, and necessity for high doses and frequent administration are a disadvantage for cephalosporins.14
Penicillins
Penicillin G is most often used in the form of procaine penicillin G that is injected i.m. or s.c. to produce long-lasting plasma concentrations for up to 24 hr. Penicillin G has a spectrum that includes gram-positive cocci, such as streptococci, anaerobic bacteria, and a few other susceptible bacteria.
Resistance is common. Ampicillin and amoxicillin, which are called the aminopenicillins, have extended spectrums that include some gram-negative cocci, but resistance also is common to these drugs . Combinations of these drugs with β-lactamase inhibitors in Unasyn (ampicillin + sulbactam) or Clavamox (amoxicillin + clavulanate) extends the spectrum to include β-lactamase-producing strains of staphylococci and gram-negative bacteria.
When penicillins are needed that have an extended-spectrum, penicillin derivatives ticarcillin and carbenicillin (carboxypenicillins) have been used. Other extended-spectrum penicillins include the ureidopenicillins (piperacillin, mezlocillin). These drugs are most often administered for treating Pseudomonas infections, although they also have activity against other gram-negative bacteria and anaerobes. Piperacillin has activity against some enterococci. Ticarcillin has been one of the most commonly used of this group for treatment of Pseudomonas infections and a few other gram-negative bacteria. (There also is an intrauterine form approved for use in horses.) Dose ranges are higher than one is accustomed for other antibiotics. Ticarcillin (Ticar® ticarcillin disodium) may be administered i.v., i.m., or s.c. at doses of 40–80 mg/kg every 6 hr. For non-i.v. administration, ticarcillin may cause pain and it is acceptable to reconstitute ticarcillin with 1% lidocaine (without epinephrine) instead of water or saline solution prior to injection.
For all the penicillin class, there is little pharmacokinetic or dosing information available for animals except the usual domestic species. Among the domestic mammals, the distribution and elimination are similar for penicillin and penicillin derivatives, therefore doses are equivalent for most animals. Oral absorption of these drugs in ruminants or horses is minimal. In non-mammals there is limited dosing information. In birds, the extended-spectrum penicillins have been used for infections resistant to other drugs, but poor oral absorption and necessity for high doses and frequent administration are a disadvantage.14 There is limited information available based on studies performed in reptiles. In general, these drugs are eliminated slowly in reptiles and dosing regimens are a reflection of the long half-lives.13
Fluoroquinolones
The fluoroquinolones include enrofloxacin (Baytril), marbofloxacin (Zeniquin), difloxacin (Dicural), and orbifloxacin (Orbax), which are currently approved for small animals (dogs, and for some, only for cats). Enrofloxacin and sarafloxacin are approved for poultry, and enrofloxacin 100 mg/ml injection is approved for cattle. There are other fluoroquinolones approved for use in human medicine (ciprofloxacin, lomefloxacin, enoxacin, ofloxacin), but except for ciprofloxacin, their used has been limited in veterinary medicine.
The fluoroquinolones have the advantage of a broad spectrum of activity that includes most gram-negative bacteria, many gram-positive bacteria (including staphylococci), Rickettsia, Chlamydia, and some Mycoplasma. Important deficiencies in the spectrum of activity include gram-positive cocci, especially enterococci (Enterococcus faecalis and Enterococcus faecium), and anaerobic bacteria.
Pharmacokinetic characteristics: Pharmacokinetic information is available for a great variety of mammals, as well as many zoo and exotic animals. Most of the data has been accumulated for enrofloxacin since it has been available for the longest time. The data on pharmacokinetics allows dosing estimates to be made for many reptiles, birds, and small mammals.13-17 In mammals, the pharmacokinetics of enrofloxacin are similar across species (i.e., half-lives of 2–6 hr, volume of distribution of 3–4 L/kg) and dosing regimens are similar. Oral absorption generally is good in animals with simple stomachs, but decreased in horses and ruminants. Although the extent of absorption may be lower in ruminants after oral administration, the half-life is longer, probably caused from delayed absorption from the rumen. In birds, there is low oral absorption and need for higher concentrations for some bacteria. Subsequently, doses cited for birds are generally higher than for mammals14 (for example 15 mg/kg/day for routine infections). In reptiles there is great variation in the pharmacokinetics, but enrofloxacin has been a valuable antibacterial drug for these animals.13,16 Our laboratory has measured pharmacokinetics and showed that half-lives for alligators, monitors, turtles, pythons, and tortoises were 55, 36, 18, 6, and 5 hr, respectively.18 Volume of distribution tended to be large as for mammals. The variation in elimination showed that all reptile species should not be treated with the same dosing intervals, and that for some, the long half-life allows for infrequent administration schedules. We also found that in the reptiles for which oral doses were administered, absorption is generally good, but that oral absorption prolonged the terminal half-life. All studies cited were done in reptiles in which temperature was kept relatively constant.
Dosage regimens: Dosages for fluoroquinolones in the approved species are flexible: enrofloxacin, 5–20 mg/kg once daily; orbifloxacin, 2.5–7.5 mg/kg once daily; marbofloxacin, 2.75–5.55 mg/kg, once daily; and difloxacin, 5–10 mg/kg once daily. The flexible dose is intended to allow for a differences in susceptibility among bacteria and increased doses for infections that may be more refractory to treatment. Gram-negative bacilli such as Escherichia coli that have a low MIC (e.g., <1.0 :g/ml) can be treated with the lowest dose in the range. Organisms that have a high MIC, (0.5–1.0 :g/ml) should be treated with the highest dose in the range. Organisms with intermediate susceptibility can be treated with moderate doses. Doses also have been published for small mammals, reptiles, and birds.13,14,17 These doses reflect the dosing requirements for susceptible bacteria in these animals based on available pharmacokinetic data and susceptibility information.
Aminoglycosides
The aminoglycosides include amikacin, gentamicin, kanamycin, and tobramycin. They have good activity against most gram-negative bacilli, including Pseudomonas aeruginosa. They have less activity against streptococci and no activity against anaerobic bacteria. Among the aminoglycosides, amikacin is the most active against Pseudomonas. Aminoglycosides can cause nephrotoxicosis, ototoxicosis, and vestibulotoxicosis. Because nephrotoxicity has been linked to persistently elevated trough concentrations, dosage regimens that increase the interval between doses has been advocated.7 The optimal bactericidal effects from these drugs is concentration-dependent. Therefore, a dose regimen that administers a single high dose, compared to two or three lower doses are equally effective. Since a single high dose is perhaps, less nephrotoxic than multiple daily doses, once-daily dosing has been adopted as routine clinical practice.7
As an example of dose regimens in mammals, gentamicin is administered at a dose of 7–10 mg/kg and amikacin at a dose of 10–15 mg/kg. Either drug is administered once daily (preferably in the morning to reduce toxicity) i.v., s.c., or i.m. To decrease emergence of resistance, and produce a more bactericidal effect, many clinicians recommend coadministration of an aminoglycoside with a β-lactam (a penicillin derivative, or cephalosporin). In addition to a broader spectrum, the β-lactam enhances bacterial penetration of an aminoglycoside.
In non-mammal species, pharmacokinetics of aminoglycosides vary greatly. Pharmacokinetics in birds do not vary significantly from mammals, but there are some differences among birds15, and some dosages have been published.14 Clearance of aminoglycosides is slow in reptiles and subsequently they are at a greater risk for nephrotoxicosis.13 For most reptiles, dose intervals are 48–96 hr.13,16 Route of injection in animals has been i.v., i.m., and s.c. In reptiles, there is a renal portal system that may affect systemic clearance for some drugs. The concern is that a drug injected i.m. in the rear leg may be cleared by the kidney before it reaches the systemic circulation. However, site of injection does not appear to affect aminoglycosides. We observer similar pharmacokinetic profiles when gentamicin was injected i.m. in the rear leg vs. front leg in turtles.19
Tetracyclines
Tetracycline antibiotics remain popular because of their wide spectrum of activity, good distribution to most body fluids, and oral absorption in most animals. Oral formulations of doxycycline have been used to treat bacterial infections as well as intracellular pathogens (Rickettsia, Chlamydia, Ehrlichia) in small animals. Injectable formulations of oxytetracycline (for example LA-200) have been used for i.m. injection because they produce prolonged plasma concentrations. Dosing information for tetracyclines is available for most domestic animals. Additional pharmacokinetic and dosing information exist for camels, wild ungulates, rabbits, fish, reptiles and birds based on pharmacokinetic studies. In reptiles, there is evidence of prolonged elimination of tetracyclines, which is probably caused by low renal clearance. In one study, the average half-life of oxytetracycline administered i.v. in alligators was 74 hr (131 hr after i.m. injection of long-acting oxytetracycline),20 compared to a range of approximately 3–10 hr in most mammals. Tetracyclines have been used to treat mycoplasma infections in reptiles and dose intervals of 72–96 hr have been used.13
Doxycycline has become a treatment of choice for chlamydial infections in birds because of its good oral absorption and efficacy. It can be dosed to pet birds by simply adding doxycycline hyclate to drinking water. When doxycycline hyclate was added to drinking water at concentrations in drinking water of 0.28 mg/ml and 0.83 mg/ml plasma concentrations were maintained high enough for susceptible organisms during a 45-day treatment. Other oral dose regimens are available.14 The oral route is preferred for doxycycline because i.m. injections cause pain and tissue irritation. Formulations of oxytetracycline have been added to drinking water of poultry to control bacterial infections.
Macrolides and Derivatives, and Lincosamides
Erythromycin is an effective drug that has been available for many years and used in a variety of domestic species. Its advantages include intracellular and tissue distribution and good safety profile. Its disadvantages include a narrow spectrum, adverse gastrointestinal effects (nausea and vomiting), poor oral absorption, short half-life, and need for frequent dosing intervals. There are new derivatives of this macrolide drug that are designed to improve therapy and produce fewer adverse reactions. Among these newer derivatives, azithromycin (Zithromax®) is the first drug in the class of azalides.21 Azalides are derived from erythromycin and the mechanism of action is similar. (Erythromycin is a 14-member ring, and azithromycin has a 15-member ring structure.) The important difference between azithromycin and erythromycin is the good oral absorption, it is better tolerated, has a much longer half-life (especially in tissues), and has a broader spectrum of activity.21
Azithromycn is active against gram-positive aerobic bacteria (staphylococci and streptococci) and anaerobes. The activity against staphylococci is not any better than erythromycin, but it has good activity against many intracellular organisms including Chlamydia, and Toxoplasma. It is also active against mycobacteria and Mycoplasma.
Azithromycin is concentrated in tissues, particularly leukocytes, macrophages and fibroblasts. The tissue concentration can be as much as 100× serum concentrations and the concentrations in leukocytes may be 200× the concentrations in serum. There has been limited use of this drug for treating infections in dogs, cats, and birds, but its popularity is increasing. Results of treatment of intracellular infections such as Toxoplasma and Mycobacterium has been encouraging in people, but not yet reported for animals.
Literature Cited
1. Nix D.E., S.D. Goodwin, C.A. Peloquin, et al. 1991. Antibiotic tissue penetration and its relevance: impact of tissue penetration on infection response. Antimicrob Agents Chemother 35: 1953–1959.
2. Pascual A. 1995. Uptake and intracellular activity of antimicrobial agents in phagocytic cells. Rev Med Microbiol 6: 228–235.
3. Lloyd D.H., A.I. Lamport, and C. Feeney. 1996. Sensitivity to antibiotics amongst cutaneous and mucosal isolates of canine pathogenic staphylococci in the UK, 1980–1996. Vet Derm 7: 171–175.
4. Lorian V. 1996. Antibiotics in Laboratory Medicine 4th Ed. Williams & Wilkins.
5. Nicolau D.P., R. Quintiliani, and C.H. Nightingale. 1995. Antibiotic kinetics and dynamics for the clinician. Med Clinics North America 79: 477–495.
6. Hyatt J.M., P.S. McKinnon, G.S. Zimmer, and J.J. Schentag. 1995. The importance of pharmacokinetic/pharmacodynamic surrogate markers to outcome. Clin Pharmacokinet 28: 143–160.
7. Freeman C.D., D.P. Nicolau, P.P. Belliveau, and C.H. Nightingale. 1997. Once-daily dosing of aminoglycosides: review and recommendations for clinical practice. J Antimicrob Chemother 39: 677.
8. Meinen J.B., J.T. McClure, and E. Rosin. 1995. Pharmacokinetics of enrofloxacin in clinically normal dogs and mice and drug pharmacodynamics in neutropenic mice with Escherichia coli and staphylococcal infections. Am J Vet Res 56: 1219–1224.
9. Lode, H., K. Borner, K., and P. Koeppe. 1998. Pharmacodynamics of fluoroquinolones. Clin Infect Dis. 1998; 27: 33–9.
10. Turnidge, J.D. 1998. The pharmacodynamics of β-lactams. Clin Infect Dis. 27: 10–22.
11. M.G. Papich, 1998. Antibacterial drug therapy: Focus on new drugs. Veterinary Clinics of North America (Small Animal Practice). 28(2): 215–231.
12. Stamper, M.A., M.G. Papich, G.A. Lewbart, S.B. May, D.D. Plummer, and M.K. Stoskopf. 1999. Pharmacokinetics of ceftazidime in loggerhead sea turtles (Caretta caretta) after single intravenous and intramuscular injections. J Zoo Wildl Med 30: 32–35.
13. Jacobson, E.R. 1999. Antimicrobial therapy in reptiles. Compend Cont Educ Pract Vet 21 (Supplement 3E): 33–48.
14. Flammer, K. 1998. Common bacterial infections and antibiotic use in companion birds. Compend Cont Educ Pract Vet 20 (Supplement 3A): 34–48.
15. Brown, S.A., and J.E. Riviere. 1991. Comparative pharmacokinetics of aminoglycosides antibiotics. J Vet Pharmacol Ther 14: 1–35.
16. Mader, D.R. 1998. Common bacterial disease and antibiotic therapy in reptiles. Compend Cont Educ Pract Vet 20 (Supplement 3A): 23–33.
17. Gobel T. 1999. Bacterial diseases and antimicrobial therapy in small mammals. Compend Cont Educ Pract Vet 21 (Supplement 3E): 5–20.
18. Papich, M.G. Pharmacokinetics of enrofloxacin in reptiles. Compend Cont Educ Pract Vet. (in press).
19. Beck, K., M. Loomis, G. Lewbart, L. Spelman, and M. Papich. 1995. Preliminary comparison of plasma concentrations of gentamicin injected into the cranial and caudal limb musculature of the Eastern box turtle (Terrapene carolina carolina). J Wildl Med 26: 265–268.
20. Helmick K.E., M.G. Papich, K.A. Vliet, et al. 1997. Preliminary kinetics of single dose intravenously administered enrofloxacin and oxytetracycline in the American alligator (Alligator mississippiensis). Proceedings of the American Association of Zoo Veterinarians. 27–28.
21. Lode H, K. Borner, P. Koeppe, and T. Schaberg. 1996. Azithromycin: review of key chemical, pharmacokinetic, and microbiological features. J Antimicrob Chemother 37 (Suppl C): 1–8.