Objectives of the Presentation
 To explain the canine genome structure and its characteristics, as revealed by the Dog Genome Project. The structure reflects the unique history of canine species and breed creation and makes the genome of the dog excellent for finding disease genes.
To explain the canine genome structure and its characteristics, as revealed by the Dog Genome Project. The structure reflects the unique history of canine species and breed creation and makes the genome of the dog excellent for finding disease genes.
To go over the development and utilization of a powerful new SNP array tool to map disease genes and traits, including some actual examples.
To discuss the status of the feline genome sequencing project.
Overview of the Issue
The Dog Genome Project which was carried out at the Broad Institute of MIT and Harvard reported a high-quality sequence of the dog genome covering ~99% of the euchromatic genome. A set of ~19,500 genes were identified and are very similar to the human genes. In addition, a map of 2.5 million single nucleotide polymorphisms (SNPs; markers useful for finding disease genes) was identified using ten different breeds and 5 canids. All of this data is available to all researchers in the world through the worldwide web.
The unique breeding history of the domestic dog provides an unparalleled opportunity to explore the genetic basis of disease susceptibility, morphological variation and behavioral traits. In-depth understanding of the canine genome is therefore important for developing an effective strategy to map a gene (genes) that is associated with certain traits or diseases. The detailed analysis of the canine genome by the Dog Genome Project using many breeds allowed us to develop a canine SNP array in collaboration with Affymetrix to effectively genotype ~27,000 SNPs covering the whole dog genome, and to design a novel 2-stage strategy to carry out whole genome association mapping. We have mapped several Mendelian traits to validate the array and our mapping method. We are currently in the process of mapping complex traits, such as cancer, that involve multiple loci. The mapping strategies, the SNP array tool and some of our results will be discussed to show how relatively straightforward the mapping process is.
The dog genome was the first to be sequenced at high coverage in Laurasiatheria, the neighboring clade to Euarchontoglires. It was a very useful addition for comparing the genomes of different mammals to better understand the genes and the regulatory regions (i.e., on-and-off switches) for the genes. To date more than 20 species of mammals, including cat, have been sequenced and are being used to determine the features in the human genome.
The first cat genome sequence was generated using low coverage (2x) to inform the human genome, which means that ~80% of the genome is captured and an in-depth analysis of the cat genome itself is difficult. This genome was sequenced by Agencourt and assembled by the Broad Institute of MIT and Harvard. Still, a preliminary analysis of synteny, repeat content, and gene content was performed by NCI, as well as SNP discovery from the sequenced Abyssinian cat. A more complete 7x genome sequence is currently being generated by Washington University in St. Louis. Further analysis of genes, as well as additional SNP discovery using multiple breeds, is planned. However, initial analysis of the genome structure suggests that a mapping array could be developed effectively with roughly 50,000 SNPs.
Additional Detail
Details of the Dog Genome Project
Detailed information of the dog genome sequence, evolution and haplotype structure can be found in a book chapter1 (in layperson's terms), as well as in a journal article2 (in more scientific terms).
Whole Genome Association Mapping In Dogs in 2 Stages
Based on the haplotype structure observed within and across breeds by the dog genome project1,2, a novel two-tiered strategy has been designed to carry out whole genome association mapping. This approach should allows one to rapidly narrow the trait-associated region to one that contains only a few genes, probably <100kb, or a single gene (Figure 1). First, a genome-wide scan for association within one breed using a ~27,000 SNP array is performed. This will permit identification of a few highly associated discrete regions (~1Mb in size). Second, because breeds with a similar phenotype are likely to share disease haplotypes, the breed used in the genome wide association study as well as small numbers of cases and controls from additional breeds that exhibit the same phenotype are used to narrow the disease-associated regions.
| Figure 1. Strategy for association mapping. | 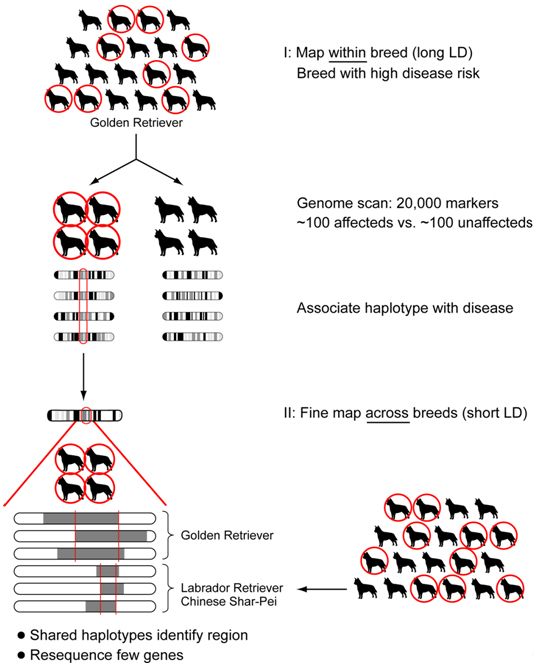
|
|
| |
Genome wide association mapping is performed in one breed, followed by fine mapping in multiple breeds. This way the long LD within breed is used for the initial association and the short LD across breeds used for localizing the mutation during fine mapping.
Summary
The domesticated dog encompasses hundreds of genetically isolated breeds and offers exceptional power for mapping genes. Due to small founder populations, specific breeds suffer increased rates of disease including cancer, epilepsy and diabetes. This population history created long linkage disequilibrium within breeds, but across breeds LD is short, suggesting a two-stage mapping strategy. An associated region is first identified by genome-wide mapping within a breed, and subsequently refined by fine-mapping across multiple breeds.
We have mapped two traits with Mendelian inheritance, white coat color (Boxers) and dermoid sinus (Rhodesian Ridgebacks), using an ~27,000 SNP array designed in collaboration with Affymetrix. For each trait, with ~10 cases and ~10 controls we identified a single associated haplotype of <1Mb containing strong candidate gene(s). For coat color, subsequent fine-mapping in two breeds narrowed the association to just 100kb containing the melanocyte-specific promoter of the gene MITF.
The real challenge lies in mapping complex traits. Promising preliminary data for osteosarcoma and hemangiosarcoma identify several loci for each using only several hundred samples. The relatively easy identification of common canine disease genes should provide insights into human health.
References
1. Wade C, Karlsson EK, Mikkelsen TS, et al. The dog genome: Sequence, evolution, and haplotype structure. In: Ostrander EA, Giger U, Lindblad-Toh K, eds. The dog and its genome. New York: Cold Spring Harbor laboratory Press, 2006;179-207
2. Lindblad-Toh K, Wade C, Mikkelsen TS, et al. Genome Sequence, Comparative Analysis and haplotype structure of the domestic dog. Nature 2005; 438:803-19