Canine hip dysplasia (CHD) is a common developmental complex trait characterized by hip joint laxity and subluxation and secondary hip osteoarthritis. To dissect the underlying genetics of this common heritable disease in dogs, an experimental pedigree was established by crossing unaffected Greyhounds and dysplastic Labrador Retrievers. Hip phenotypes measured at 8 months and analyzed included the distraction index, the dorsolateral subluxation score and the Norberg angle. A first genome-wide screen on 152 dogs from this 3 generation pedigree was completed with 250 microsatellites at the NHLBI Mammalian Genotyping Service, Marshfield WI. We reported (Todhunter et al, 2005 Mammalian Genome) identification of putative QTL for canine hip dysplasia (p<0.05, chromosome-wide) on CFA04, 09, 10, 11 (p<0.01), 16, 20, 22, 25, 29 (p<0.01, LOD score 2.8), 30, 35, and 37.
| Figure 1. | 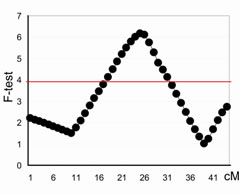
Profile plot of F-test statistics (vertical axis) of the QTL underlying the NA on chromosome 29. The peak represents position of the QTL in cM on the X axis across the chromosome. The threshold at a chromosome-wide significance level of 0.05 is drawn as a solid red line across the plot. |
|
| |
To improve the marker coverage and to provide a better resolution in defining the QTL location, a second genome-wide screen was undertaken with 323 additional markers from the Minimal Screen Set 2 (MSS2). Twenty-five chromosomes were genotyped and integrated with the previous screen, resulting in 460 unique marker genotypes. Genotyping is continuing on the remaining 14 chromosomes. Interval mapping analysis was performed using the combined backcross/F2 design module from QTL Express, a web-based software which regresses identical-by-descent marker probabilities on the phenotypes. Estimates were obtained for the additive and dominance effect of the putative QTL at each location across the chromosome. The location giving the highest F ratio statistic was considered to be the best estimate for the position of the QTL. Chromosome-wide significance thresholds for each trait were determined empirically by permuting the marker data; the threshold was obtained from 500 permutations. Putative QTL were identified on 11 out of the 25 chromosomes tested with the full set of markers. The QTL detected on chromosomes 11 and 29 (Figure 1) remain with the highest LOD scores and are the focus of initial fine mapping.
To further validate these results, 130 purebred Labrador Retrievers were genotyped using a revised Mammalian Genotyping Service set of 249 markers at Marshfield Medical Research Foundation. Genotyping on this Labrador Retriever population with the MSS2 markers is completed on 25 chromosomes. QTL mapping is performed using a new module of QTL Express designed to analyze data from purebred populations with complex pedigrees using a variance-based approach. Fine mapping of the validated QTL will be undertaken using SNP haplotypes on the crossbred pedigree, purebred pedigree and also other breeds afflicted with the disease.
Identifying QTL which either confer protection or increases the risk of CHD should provide dog breeders with the tools needed to implement an effective selection program to reduce the frequency of CHD and veterinarians and dog owners with effective methods to identify dogs at risk of developing CHD early in life, when preventive measures can be implemented. Identifying the mutation(s) contributing to CHD will increases our understanding of CHD and should open up new and more effective therapies for this orthopedic disease of dogs.
Support: Morris Animal Foundation; Collaborative Grant Program, College of Veterinary Medicine and Biotechnology Center for Advanced Technology, Cornell University; American Border Collie Association; Van Sloun Foundation; Masterfoods Inc; Mammalian Genotyping Service